13.15 A Re-Sampling Approach
Re-sampling approach to hypothesis testing using the Khan dataset.
attach(Khan)
x <- rbind(xtrain, xtest)
y <- c(as.numeric(ytrain), as.numeric(ytest))
dim(x)## [1] 83 2308table(y)## y
## 1 2 3 4
## 11 29 18 25There are four classes of cancer.
For each gene, we compare the mean expression in the second class (rhabdomyosarcoma) to the mean expression in the fourth class (Burkitt’s lymphoma).
x <- as.matrix(x)
x1 <- x[which(y == 2), ]
x2 <- x[which(y == 4), ]
n1 <- nrow(x1)
n2 <- nrow(x2)
t.out <- t.test(x1[, 11], x2[, 11], var.equal = TRUE)
TT <- t.out$statistic
TT## t
## -2.093633t.out$p.value## [1] 0.04118644Instead of using this theoretical null distribution, we can randomly split the 54 patients into two groups of 29 and 25, and compute a new test statistic.
Repeating this process 10,000 times:
set.seed(1)
B <- 10000
Tbs <- rep(NA, B)
for (b in 1:B) {
dat <- sample(c(x1[, 11], x2[, 11]))
Tbs[b] <- t.test(dat[1:n1], dat[(n1 + 1):(n1 + n2)],
var.equal = TRUE
)$statistic
}
mean((abs(Tbs) >= abs(TT)))## [1] 0.0416This fraction, \(0.0416\), is our re-sampling-based \(p\)-value. It is almost identical to the \(p\)-value of \(0.0412\) obtained using the theoretical null distribution.
A histogram of the re-sampling-based test statistics:
hist(Tbs, breaks = 100, xlim = c(-4.2, 4.2), main = "",
xlab = "Null Distribution of Test Statistic", col = 7)
lines(seq(-4.2, 4.2, len = 1000),
dt(seq(-4.2, 4.2, len = 1000),
df = (n1 + n2 - 2)
) * 1000, col = 2, lwd = 3)
abline(v = TT, col = 4, lwd = 2)
text(TT + 0.5, 350, paste("T = ", round(TT, 4), sep = ""),
col = 4)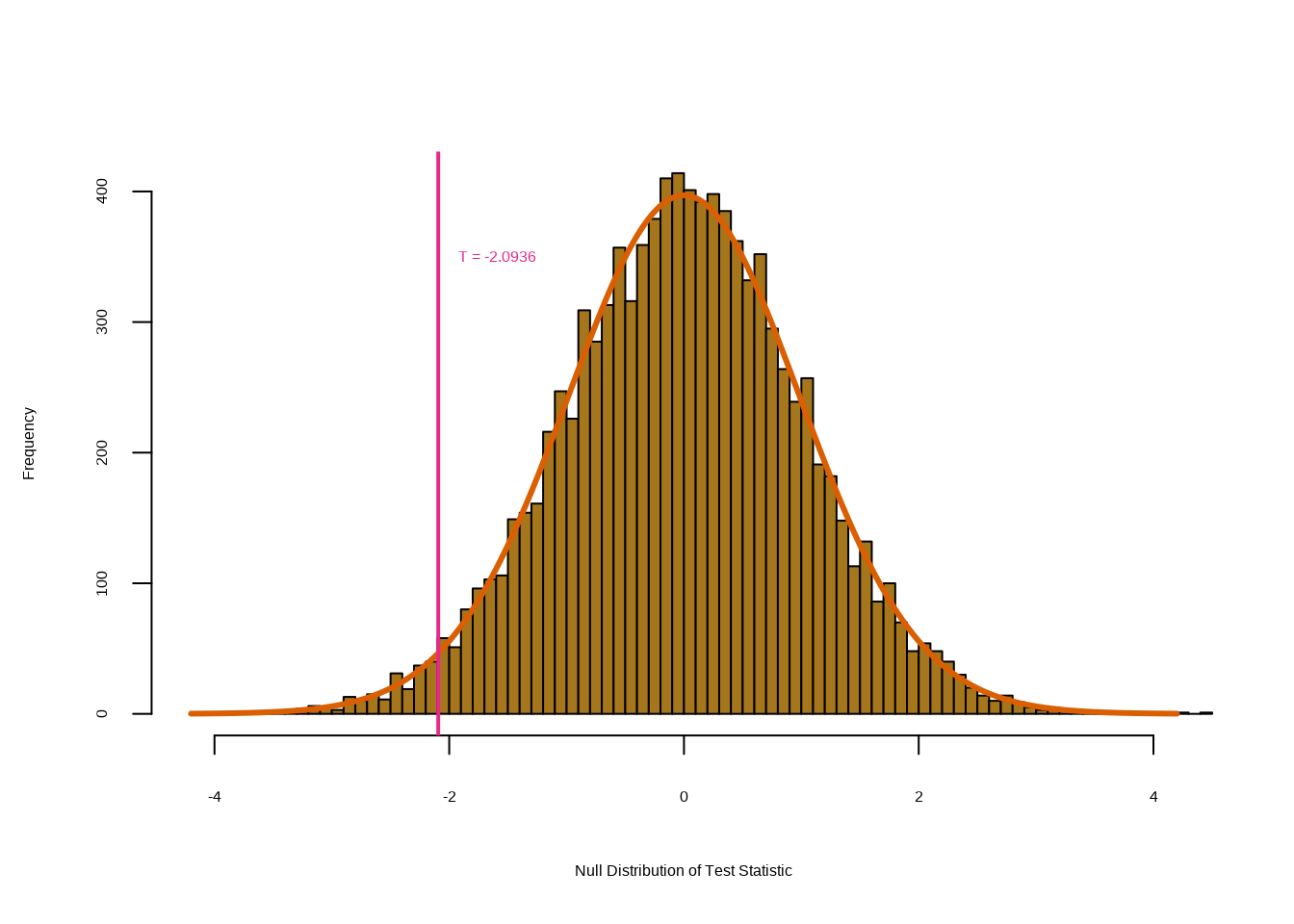
For each gene, we first compute the observed test statistic,
m <- 100
B<-1000
set.seed(1)
index <- sample(ncol(x1), m)
Ts <- rep(NA, m)
Ts.star <- matrix(NA, ncol = m, nrow = B)
for (j in 1:m) {
k <- index[j]
Ts[j] <- t.test(x1[, k], x2[, k],
var.equal = TRUE
)$statistic
for (b in 1:B) {
dat <- sample(c(x1[, k], x2[, k]))
Ts.star[b, j] <- t.test(dat[1:n1],
dat[(n1 + 1):(n1 + n2)], var.equal = TRUE
)$statistic
}
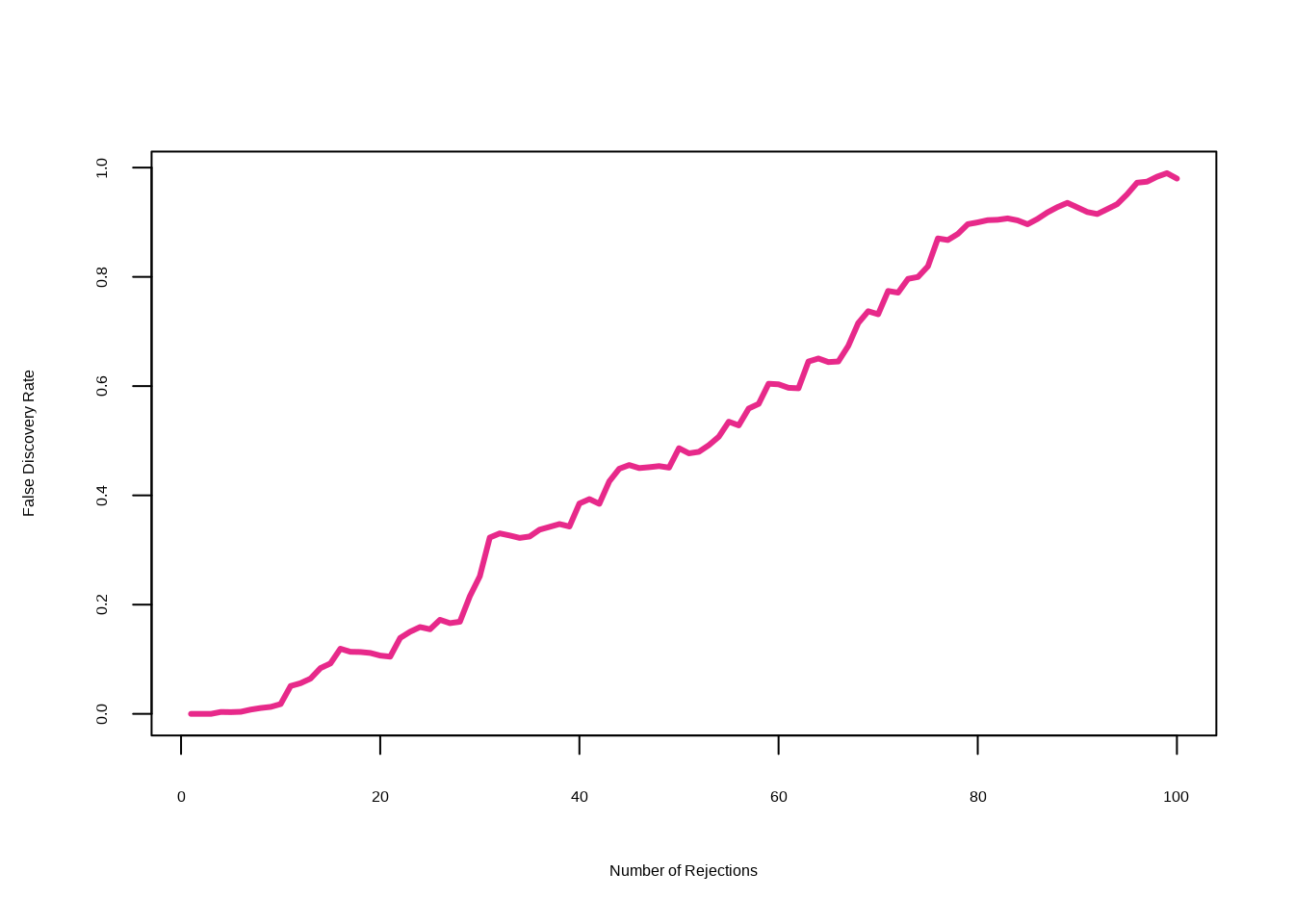
}Compute a number of rejected null hypotheses \(R\).
cs <- sort(abs(Ts))
FDRs <- Rs <- Vs <- rep(NA, m)
for (j in 1:m) {
R <- sum(abs(Ts) >= cs[j])
V <- sum(abs(Ts.star) >= cs[j]) / B
Rs[j] <- R
Vs[j] <- V
FDRs[j] <- V / R
}The variable index is needed here since we restricted our analysis to just \(100\) randomly-selected genes.
max(Rs[FDRs <= .1])## [1] 15sort(index[abs(Ts) >= min(cs[FDRs < .1])])## [1] 29 465 501 554 573 729 733 1301 1317 1640 1646 1706 1799 1942 2159max(Rs[FDRs <= .2])## [1] 28sort(index[abs(Ts) >= min(cs[FDRs < .2])])## [1] 29 40 287 361 369 465 501 554 573 679 729 733 990 1069 1073
## [16] 1301 1317 1414 1639 1640 1646 1706 1799 1826 1942 1974 2087 2159plot(Rs, FDRs, xlab = "Number of Rejections", type = "l",
ylab = "False Discovery Rate", col = 4, lwd = 3)