6.1 Genomic Ranges
6.1.1 GRanges objects
Helpful GenomicRanges Walkthrough
Tidy Manipulation of GRanges Objects
GRanges Object: data table like, can be manipulated like a data table for the most part
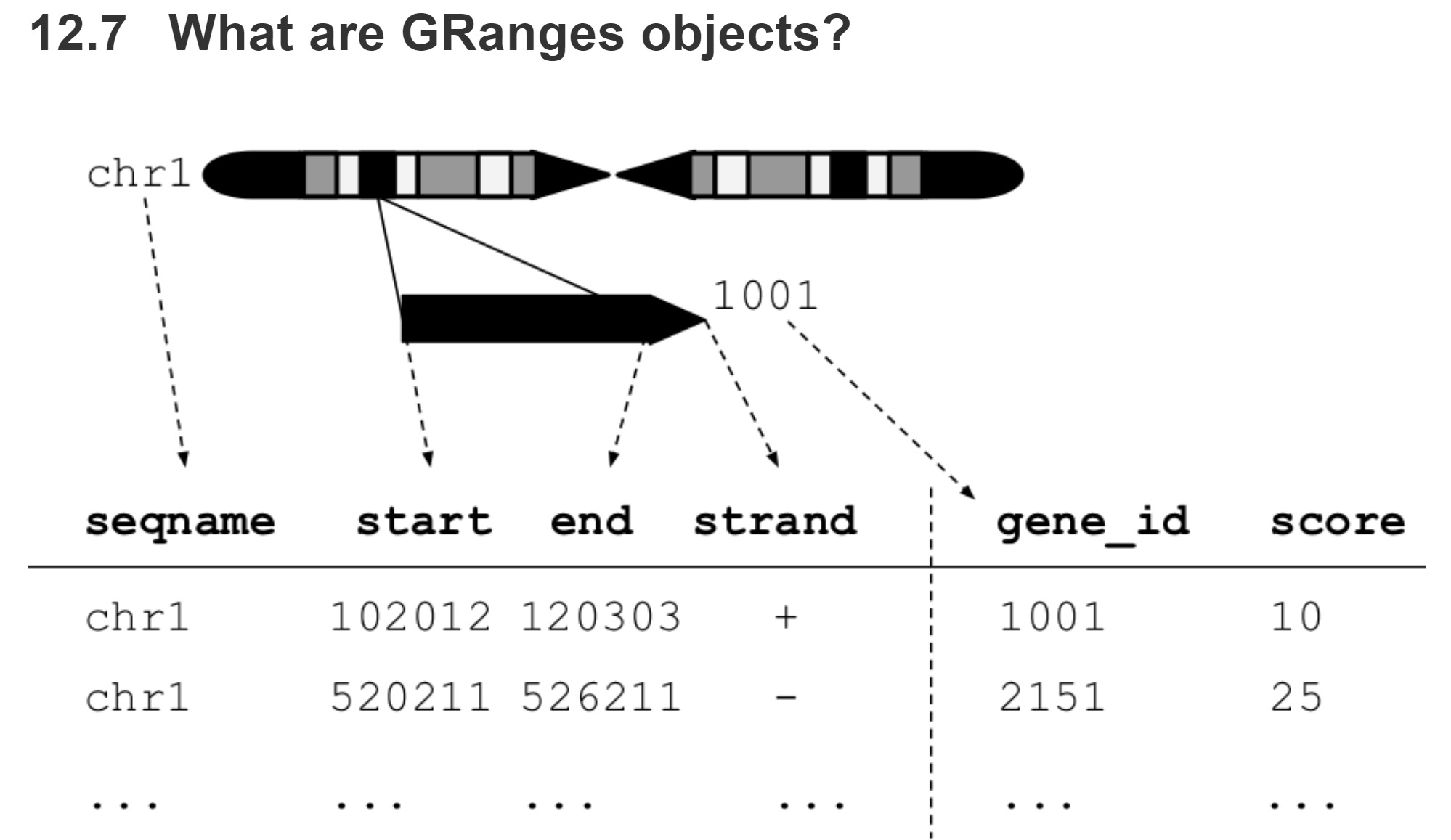
GRanges
- Three Primary Components of a GRanges Object
- SeqNames (vector)
- Ranges (IRanges Object)
- Strand (vector)
- Metadata
- Added as vectors, can be nearly anything (mcols)
## Loading required package: stats4## Loading required package: BiocGenerics##
## Attaching package: 'BiocGenerics'## The following object is masked from 'package:kernlab':
##
## type## The following object is masked from 'package:pROC':
##
## var## The following objects are masked from 'package:mosaic':
##
## counts, IQR, sd, var## The following objects are masked from 'package:dplyr':
##
## combine, intersect, setdiff, union## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, aperm, append, as.data.frame, basename, cbind,
## colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
## get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
## match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
## Position, rank, rbind, Reduce, rownames, sapply, setdiff, sort,
## table, tapply, union, unique, unsplit, which.max, which.min## Loading required package: S4Vectors##
## Attaching package: 'S4Vectors'## The following objects are masked from 'package:dplyr':
##
## first, rename## The following objects are masked from 'package:Matrix':
##
## expand, unname## The following object is masked from 'package:utils':
##
## findMatches## The following objects are masked from 'package:base':
##
## expand.grid, I, unname## Loading required package: IRanges##
## Attaching package: 'IRanges'## The following object is masked from 'package:xgboost':
##
## slice## The following object is masked from 'package:mosaic':
##
## mid## The following objects are masked from 'package:dplyr':
##
## collapse, desc, slice## Loading required package: GenomeInfoDbgr=GRanges(seqnames=c("chr1","chr2","chr2"),
ranges=IRanges(start=c(50,150,200),
end=c(100,200,300)),
strand=c("+","-","-")
)
gr## GRanges object with 3 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 50-100 +
## [2] chr2 150-200 -
## [3] chr2 200-300 -
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## [1] 50 150 200## [1] 100 200 300## [1] 51 51 101## GRanges object with 2 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 50-100 +
## [2] chr2 150-300 -
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## GRanges object with 2 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 50-100 +
## [2] chr2 150-200 -
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths##
## Attaching package: 'plyranges'## The following object is masked from 'package:IRanges':
##
## slice## The following object is masked from 'package:xgboost':
##
## slice## The following objects are masked from 'package:dplyr':
##
## between, n, n_distinct## The following object is masked from 'package:stats':
##
## filterdf <- data.frame(seqnames = c("chr1", "chr2", "chr2", "chr1", "chr2"),
start = 50:54,
width = 5)
df$strand <- c("+", "+", "-", "-", "*")
grdf <- as_granges(df)
grdf %>%
filter(strand == "-")## GRanges object with 2 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr2 52-56 -
## [2] chr1 53-57 -
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## GRanges object with 3 ranges and 3 metadata columns:
## seqnames ranges strand | name2 score2 name3
## <Rle> <IRanges> <Rle> | <character> <numeric> <character>
## [1] chr1 50-100 + | pax6 1 A
## [2] chr2 150-200 - | meis1 2 C
## [3] chr2 200-300 - | zic4 3 B
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsGRangeList – multiple GRanges objects in a list
gr1 <- GRanges(
seqnames = "chr2",
ranges = IRanges(103, 106),
strand = "+",
score = 5L, GC = 0.45)
gr2 <- GRanges(
seqnames = c("chr1", "chr1"),
ranges = IRanges(c(107, 113), width = 3),
strand = c("+", "-"),
score = 3:4, GC = c(0.3, 0.5))
grl <- GRangesList("txA" = gr1, "txB" = gr2)
grl## GRangesList object of length 2:
## $txA
## GRanges object with 1 range and 2 metadata columns:
## seqnames ranges strand | score GC
## <Rle> <IRanges> <Rle> | <integer> <numeric>
## [1] chr2 103-106 + | 5 0.45
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $txB
## GRanges object with 2 ranges and 2 metadata columns:
## seqnames ranges strand | score GC
## <Rle> <IRanges> <Rle> | <integer> <numeric>
## [1] chr1 107-109 + | 3 0.3
## [2] chr1 113-115 - | 4 0.5
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths6.1.2 Getting genomic data into R as a table
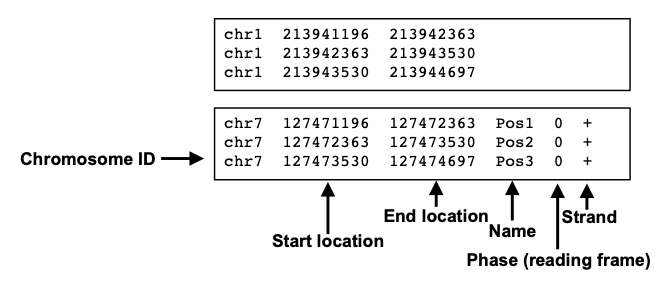
.bed file
# .bed
filePath=system.file("extdata",
"cpgi.hg19.chr21.bed",
package="compGenomRData")
cpgi.df = read.table(filePath, header = FALSE,
stringsAsFactors=FALSE)
genomation::readBed()
# .gff -- similar to .bed
genomation::gffToGranges()
rtracklayer::import.gff()
# BAM/SAM -- location of aligned reads on chromosome/genome
GenomicAlignments::readGAlignments()
Rsamtools::scanBam()
# BigWig -- scores associated w/ genomic intervals -- HUGE files so only import what you need!
rtracklayer::import.bw()
# Tabix/BCF -- lists storing genomic variations (SNPs, Indels, etc)
Rsamtools::scanTabix()
Rsamtools::scanBcf()
# Generic
genomation::readGeneric()6.1.3 Manipulating GRange Objects
intra-range: manipulate ranges within object INDEPENDENT of other ranges in object
## GRanges object with 3 ranges and 3 metadata columns:
## seqnames ranges strand | name2 score2 name3
## <Rle> <IRanges> <Rle> | <character> <numeric> <character>
## [1] chr1 40-49 + | pax6 1 A
## [2] chr2 201-210 - | meis1 2 C
## [3] chr2 301-310 - | zic4 3 B
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## GRanges object with 3 ranges and 3 metadata columns:
## seqnames ranges strand | name2 score2 name3
## <Rle> <IRanges> <Rle> | <character> <numeric> <character>
## [1] chr1 101-110 + | pax6 1 A
## [2] chr2 140-149 - | meis1 2 C
## [3] chr2 190-199 - | zic4 3 B
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## GRanges object with 3 ranges and 3 metadata columns:
## seqnames ranges strand | name2 score2 name3
## <Rle> <IRanges> <Rle> | <character> <numeric> <character>
## [1] chr1 60-110 + | pax6 1 A
## [2] chr2 160-210 - | meis1 2 C
## [3] chr2 210-310 - | zic4 3 B
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## GRanges object with 3 ranges and 3 metadata columns:
## seqnames ranges strand | name2 score2 name3
## <Rle> <IRanges> <Rle> | <character> <numeric> <character>
## [1] chr1 50-64 + | pax6 1 A
## [2] chr2 186-200 - | meis1 2 C
## [3] chr2 286-300 - | zic4 3 B
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsinter-range: comparison of ranges within a single object
## GRanges object with 2 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 50-100 +
## [2] chr2 150-300 -
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## GRanges object with 2 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 1-49 +
## [2] chr2 1-149 -
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## GRanges object with 4 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 50-100 +
## [2] chr2 150-199 -
## [3] chr2 200 -
## [4] chr2 201-300 -
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## RleList of length 2
## $chr1
## integer-Rle of length 100 with 2 runs
## Lengths: 49 51
## Values : 0 1
##
## $chr2
## integer-Rle of length 300 with 4 runs
## Lengths: 149 50 1 100
## Values : 0 1 2 1between-range: calculate ranges between DIFFERENT GRanges objects
## GRanges object with 1 range and 3 metadata columns:
## seqnames ranges strand | name2 score2 name3
## <Rle> <IRanges> <Rle> | <character> <numeric> <character>
## [1] chr1 50-100 + | pax6 1 A
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths## Hits object with 1 hit and 0 metadata columns:
## queryHits subjectHits
## <integer> <integer>
## [1] 1 1
## -------
## queryLength: 3 / subjectLength: 5## [1] 1 0 0## [1] 1 5 5## Hits object with 3 hits and 1 metadata column:
## queryHits subjectHits | distance
## <integer> <integer> | <integer>
## [1] 1 1 | 0
## [2] 2 5 | 91
## [3] 3 5 | 141
## -------
## queryLength: 3 / subjectLength: 5