7.1 .fasta vs .fastq
.fasta file starts with >
 Fastq file has four components
Fastq file has four components
 Fastq scores depend on the sequencing platform
Fastq scores depend on the sequencing platform
 ## Quality check with Rqc
## Quality check with Rqc
library(Rqc)
folder = system.file(package="ShortRead", "extdata/E-MTAB-1147")
# feeds fastq.qz files to quality check function
qcRes=rqc(path = folder, pattern = ".fastq.gz")# try with long read data... not nearly as useful
qcResC=rqc(path = "example_files/example_fastqgz", pattern = ".fastq.gz")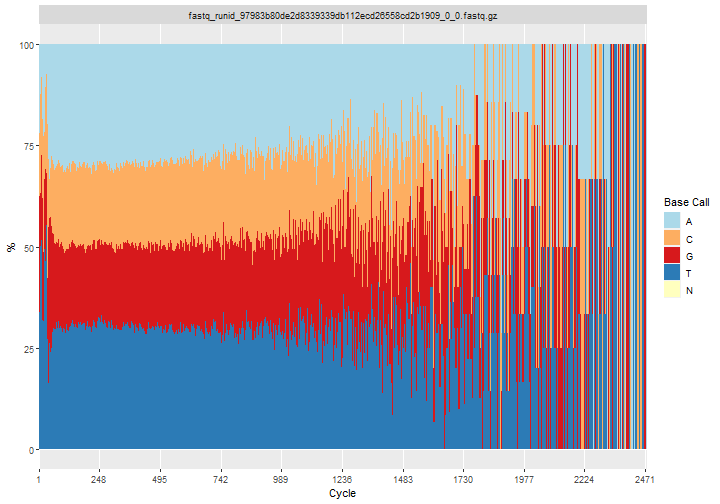
long read analysis
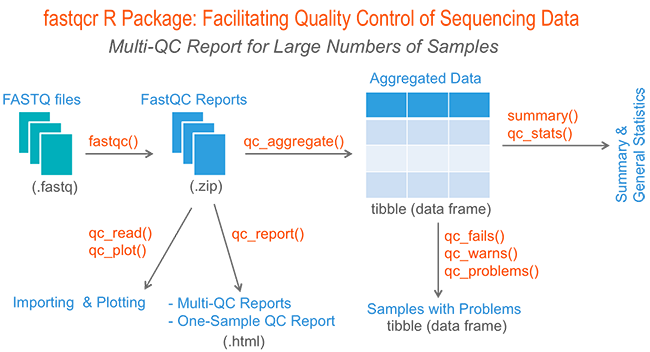
An alternative: fastqcr (CRNA not Bioconductor) for lots of fastq files! UNIX/MAC ONLY